
Background and overview[1]
Sitagliptin, chemical name (2R)-4-oxo-4-[3-trifluoromethyl-5,6-dihydro[1,2,4]triazole[4 , 3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)-butan-2-amine, which is a dipeptide developed by Merck, USA The first product of DPP-VI (DPP-VI) inhibitor. In October 2006, its phosphate monohydrate was approved by the US FDA as the first dipeptidyl peptidase-VI (DPP-VI) inhibitor for clinical use in the treatment of type 2 diabetes. The advantage of this drug is that it has few adverse reactions, low risk of hypoglycemia and does not cause weight gain. In the prior art, sitagliptin impurity 13 is an intermediate compound for the synthesis of sitagliptin. After further generating an enamine compound, a chiral amine intermediate can be further prepared through an asymmetric catalysis method. However, the yield is relatively low. Low. Moreover, after repeating the technology of the patent, it was found that the reaction could not proceed at all.
Preparation[1]
Sitagliptin Impurity 13 is prepared as follows:
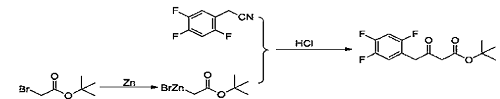
1) Add tetrahydrofuran (450g) and zinc powder (46.09g, 704mmol) to a 1000ml three-necked flask, and start magnetic stirring. Trimethylsilyl chloride (3.8g, 35mmol) was added dropwise under nitrogen protection, the temperature was raised to 40-45°C, and stirred. Under nitrogen protection, add dropwise a mixed solution of tert-butyl bromoacetate (78.0g, 400mmol) and tetrahydrofuran (150g) at 40-45°C until the drops are completed. Add 2,4,5-trifluorophenyl acetonitrile (40.0g, 234mmol) to the reaction solution under nitrogen protection, and react at 40-45°C. After the reaction was completed, filter and rinse the filter cake with tetrahydrofuran (100g). 10% hydrochloric acid aqueous solution (200 mL) was added dropwise to the filtrate and stirred. Distill under reduced pressure at 30 to 35°C. Stop distillation when there is no fraction in the condenser tube. Dichloromethane (300 mL) was added to the residue and stirred. Separate layers, remove the organic phase and discard the aqueous phase. The organic phase was washed with 10% sodium chloride aqueous solution and dried over anhydrous sodium sulfate. Filter, and the filtrate is concentrated to dryness under reduced pressure to obtain an orange oil (64.40 g, 223.4 mmol), which is the ketone tert-butyl ester compound, with a molar yield of 95.5% and a purity of 99%.
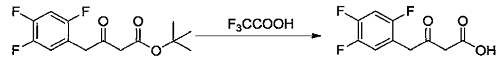
2) Add methylene chloride (50ml) and the ketone tert-butyl ester compound (2.7g, 9.37mmol) prepared in Example 7 into a 100ml single-necked flask, stir and dissolve. Trifluoroacetic acid (5.0 ml, 67.31 mmol) was added at 10-15°C, then the temperature was raised to 25-30°C and the reaction was stirred. When the reaction is complete, add water and stir. Separate layers, remove the organic phase and discard the aqueous phase. The organic phase was washed with 5% sodium chloride aqueous solution and concentrated to dryness under reduced pressure at 30-40°C to obtain an off-white solid (2.0g, 8.61mmol), which is a ketoacid compound, with a molar yield of 91.94%.
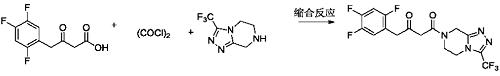
3) Add methylene chloride (100ml) and the ketoacid compound (10.0g, 43.07mmol) prepared in Example 8 into a 250ml three-necked flask, stir and dissolve. Add oxalyl chloride (6.56g, 51.68mmol) dropwise at 10-15°C. After the dropwise addition is completed, raise the temperature to 30-35°C and stir for reaction. The reaction is complete. Concentrate to dryness under reduced pressure at 30-40°C to obtain a light yellow liquid (10.91g, 43.64mmol). Add methylene chloride (100ml), triethylamine (5.79g, 57.24mmol) and the above-mentioned light yellow liquid to a 250ml three-necked flask. Yellow liquid (10.0g, 52.04mmol), stir to dissolve. Cool the temperature to 0-5°C and add dropwise the free base (13.0g, 52.04mmol)/methylene chloride (20ml) solution of the compound of formula a until the dropwise addition is completed. After the dropwise addition is completed, the temperature is raised to 10-15°C and the reaction is stirred. When the reaction is complete, add water and stir. Separate layers, remove the organic phase and discard the aqueous phase. The organic phase was washed with 5% sodium chloride aqueous solution and concentrated to dryness under reduced pressure at 30-40°C to obtain an off-white solid sitagliptin impurity 13 (19.24g, 47.36mmol), a ketoamide compound, with a molar yield of 91%. .
Main reference materials
[1] CN201711403311.2 Preparation method of sitagliptin intermediate
 微信扫一扫打赏
微信扫一扫打赏

